[Ans] For
which of the following conditions would the
"plateau" occur sooner in the face of the same
infusion rate? (hint: think bathtub!)
a) very rapid elimination?
b) very slow elimination?
[Ans] Will the
concentration of a drug plateau if it is eliminated at a
constant mass per minute, i.e., its elimination rate
could be described as 10 mcg/min?
[Ans] What could
one do if the time-to-plateau were too long for the
therapeutic need?
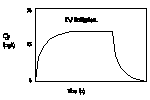
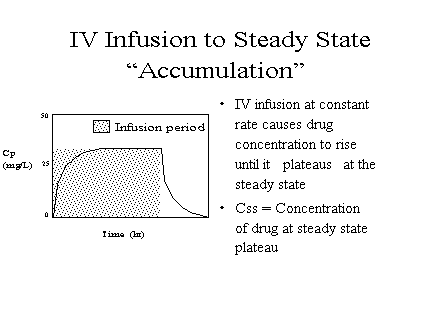
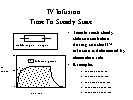
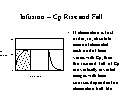
Time to
plateau: The rate at
which steady state is approached when repeated equal
doses or an intravenous infusion is given depends in
the simplest case on the elimination rate. Steady
state is approached faster when elimination is faster.
Drug "accumulates" until the
concentration gets high enough to make elimination equal
input.
[full] [icon] Figure: Intravenous
infusion an an example of "accumulation"
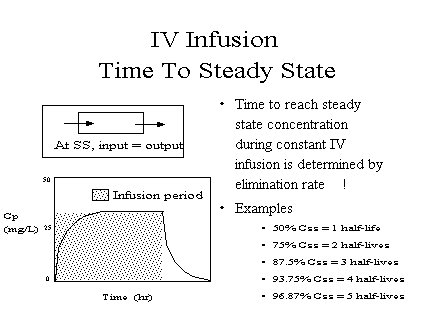
A fact you should remember is that steady state is
approached within 3 to 5 elmination half-lives (88% to
97%)
[full] [icon] Figure: Time to steady
state on intravenous infusion
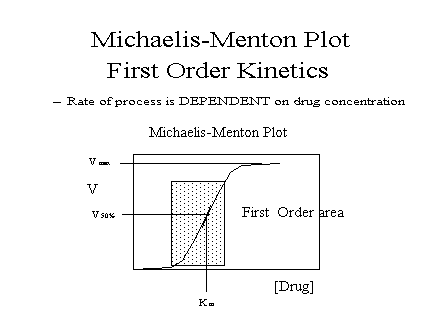
First/zero order: Drug
must follow "First-Order Kinetics" These
principles do not apply to drugs following
"Zero-Order Kinetics." These concepts apply
only when elimination is "first order", that
is, dependent on the plasma drug concentration. See
accompanying Michaelis-Menton plot of drug elimination
rate versus concentration.
[full] [icon] Figure: Michaelis-Menten
plot
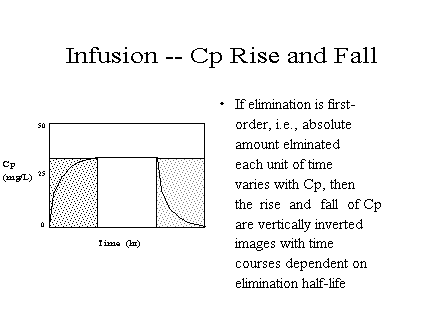
Note: the final curve after infusion is stopped is the
inverted-image" of the initial rise. Both shapes
depend on the same processes -- those involved in
eliminating the drug.
[full] [icon] Figure: Rise on
start and Fall on cessation are inverted images
Loading Dose: One can give a
first dose much larger than the maintenance dose
or, in the case of an infusion, one could give a rapid,
injection followed by the planned rate of infusion. This
concept is similar to induction and maintenance
in inhalation anesthesia. More on this later.
Assume you decide to treat 5 expensive fish in a 50 L
tank with miraclemycin and that the desired
final concentration is 10 mg/L.
How much drug (in mg) should be added?
How much drug should be given to a patient that weighs 75
kg and has a body water content of 50 L (assume uniform
distribution of drug). Desired concentrationis 10 mg/L.
How much drug (in mg) should be added?
Can you see the similarity in these examples? You have a
known volume of fluid in which you want to have a
specific concentration of drug.
Amount of Drug = Desired Conc * Volume
{mg = (mg/L) * L}
In this case,
Amount of drug = dose
Desired concentration = target concentration
(mean or average) in sampled fluid
Volume = the volume of sampled fluid in
which the drug is distributed
These cases assume that no drug has left the tank
or body, i.e.,they are values taken at the instant
drug was administered. This time is called zero-time.
They further assume that no drug was lost during
that instant
How REALISTIC are these assumptions? We will see that at
the first level of approximation and given a few
qualifiers that you are to know, they are not bad.
Loading Dose (DL) = a dose of
drug sufficient to produce a plasma conentration that
would fall within the therapeutic window after only one
or very few doses over a very short interval. It is
larger than the dose rate needed to maintain the
concentration within the window and would produce toxic
concentrations if given in repeated doses.
Maintenance Dose (DM) = The dose needed
to maintain the concentrationwithin the
therapeutic window when given repeatedly at a constant
interval
Information needed to calculate a DL
Volume of "tank" (Volume of distribution,
usually in L or L/Kg)
from literature
Desired concentration
from literature
Knowledge of assumptions
absorption very rapid relative to elimination
volume of tank is in terms of the sampled
fluid (i.e., plasma or serum) because this
is the only concentration you know by
direct measurement.
one must use the same terms throughout
calculations
mg / L; mcg/L; mcg/mL, etc.
Note that mg/L and mcg/mL are the same!
total drug vs unbound drug Usually use total drug concentration
due to cost-effectiveness
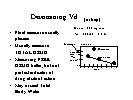
Finding Vd
Look it up!
Simple experiments
Conceptual approach given here. Actual
determinations are much more complex and by
different methods to get more accurate values
Vd Experiment #1 (closed system)
MEASURE DILUTION!
Add 500 mg of drug to ____ L of water in a beaker
Measured drug concentration after mixing is 5
mg/L
What is the volume of distribution?
Earlier formula rearranged!
Vd = Dose / Measured Conc
{L = mg / (mg/L) }
"Vd" is equivalent to
"volume"
"dose" is equivalent to "amount of
drug"
"measured" is equivalent to "desired"
{Note: in "real"
pharmacokinetics, one would use
much more complex analyses to find what is known
as the volume of distribution at steady state (Vd(ss))}
Plot the data on semi-logarithmic graph
paper with labels
(Keep this graph for later use!)
Note that on a 2 or 3-cycle sheet of
paper, each "1' could represent 0.1,
1.0, 10.0, and 100.0! For this
experiment, make the top-most
"1" = 100 mg/L.
Plot the data and connect the data
points.
Draw light horizontal lines to each data
point as you did previously
What difference do you note? The vertical
distances between each of the data points
are now the same!
Semi-log paper represents a
"geometric progression" where
each multiple is distributed evenly,
i.e., 1, 2, 4, 8, 16, 32, 64, 128, ...,
n*2 are distributed evenly. OR 1, 10,
100, 1,000, ..., n*10 are distributed
evenly on one of the axes.
By the same token, if we start with 100
ug/ml and cut the concentration by half
to 50 and then again to 25, the vertical
distances are the same. Each FRACTIONAL
change of 0.5 gets the same vertical
distance.
Can you make a meaningful extrapolation
of these data to "zero hours"?
YES! [Excel plot]
What is the concentration at the
"zero hours" intercept?
What is the volume into which the drug
APPEARS to have distributed? That is, how
much plasma-equivalent is required
to explain the dilution?
How does this compare to the weight of the
patient?
If body water is approximately 70% of body
weight, how can a drug appear to have distributed
into 2 L/Kg of body weight?
Hint: Think about what fluid we are measuring and
our assumption of uniform distribution? Are these
data consistent with the notion that miraclemycin
distributes in such a way that the concentration
outside the plasma is higher than that in the
plasma?
How can one use an estimate of Vd to calculate a
loading dose?
What is the "pharmacokinetic
usefulness" of the Vd term?
Definition: volume of plasma/serum (Vd)
required to explain the plasma/serum concentration (C)
when there is a known amount (A) of drug in the body. The
amount (A) is equivalent to the dose (D) at zero time
after administration of a single dose. For many of our
purposes, this would equate to an initial dose, or a
loading dose (DL)
Note the dimensions of the terms below. Vd can be in L or
in L/Kg. The former would be the Vd for the entire
patient, whereas the term "L/Kg" is a way of
normalizing the Vd to account for differences in body
mass.
Vd = D / Measured Conc {L = mg / (mg/L)}
One can rearrange the formula for various uses. To find a
loading dose (DL),
DL = Vd * Desired Conc {mg = L * (mg/L)}
A frequent use is in formulae to replace the
"initial concentration" (C0),
Predicted Conc = DL /Vd { mg/L = mg / L }
The easiest way to remember these is to use the most
obvious form of the equation, the one above where , and then do simple rearrangements
as needed. ALWAYS CHECK the TERMS to be
sure you are correct.
The Vd is not a specific anatomic space
Vd changes in various conditions, e.g., with age,
pregnancy, disease, levels of protein binding of drug,
fat content of the animal, etc.
The simplest conceptual way to determine Vd is to
administer a dose of drug, measure concentration over
time and extrapolate the concentrations to
"zero-time" to find the initial concentration.
Then use the known dose (DL) and concentration (C0) to calculate Vd.
[full] [icon] Figure: Example of using
a simple plot to determine Vd
Clearance is a measure of how rapidly the organ(s) of
elimination "clear" drug from the plasma.
Clearance (CL) can be defined as the
volume of plasma completely freed of drug per unit of
time (e.g., L/h). It is a "virtual" volume that
depends on two major factors for any specific organ of
clearance:
blood (plasma) flow to the organ
fraction of drug extracted from the blood
(plasma)
Extraction ratio
Extraction ratio (ER) is the
fraction of drug removed in a single pass through
the organ of elimination. It can be measured in a
single organ by sampling drug concentration in
the arterial blood into (Cin) and venous blood
from (Cout) the organ.
ER = [Cin - Cout] / Cin
Thus if the arterial blood contains 15 mg/L of
drug "A" and venous blood from the
organ contains 9 mg/L, the ER is (15-9)/15 = 6/15
= 0.4.
In the preceding example, an ER of 0.4 means that
40% of the drug in the plasma is removed as it
passes through the organ.
Extraction ratios for drugs in various organs can
range from 0 to 1.0 and may vary depending on the
functional capacity of the organ and whether or
not the drug concentration is in the
"first-order" range of concentrations.
Disease conditions and other drugs can change the
ER of a drug
Extraction ratios can also vary with blood flow
if it causes the capacity of the organ to be
exceeded.
Blood flow (often symbolized by Q with a
dot on top, but not in these notes because of limitations
in the formatting language, html) can be expressed as
mL/min, L/h, etc.
If an organ of elimination is capable of removing
all drug in a single pass, i.e., ER = 1.0, then
changes in blood flow to the organ can be very
important in determining the elimination rate
because blood flow is the rate limiting factor.
Conversely, if ER is very low, changes in blood
flow are less important.
Renal blood flow can vary widely. "Renal
shut-down" can dramatically slow elimination
of a drug as can slowed renal blood flow due to
hypovolemic conditions. For this reason, it is
common in clinical medicine to administer fluids
to keep renal perfusion high to aid in
elimination of some drugs.
Single organ clearance --
CL = ER * Flow [L/h = frcn x L/h]
Notice that there are no "mass" terms in the
final CL formula. It is the "virtual"
volume (in liters) of plasma completely cleared of
drug per hour in this case. When the ER is 0.5, the
concentration in "twice the CL volume" is
lowered by 50%.
Amount eliminated -- So,
how does one find the "amount" of drug actually
eliminated during a specific time interval from these
computations? Awareness of the difference between
"amount" and "fraction" is crucial to
understanding clinical pharmacokinetics. Be sure to pay
attention to which of these is being used in various
equations and terms. In general, when a
pharmacokineticist speaks of a "rate", one is
referring to a fraction/time.
One finds the "amount" eliminated at
any instant in time (e.g., one minute) by
multiplying the average drug concentration in the
plasma "during that minute" by the CL.
where Cin(t) represents the average
concentration of drug in the arterial plasma
during the time interval denoted by
"t".
Amount changes: One
should note that this "amount"
changes constantly as the concentration of
drug in the body changes (increasing while amount
absorbed is greater than that eliminated and
decreasing when it is less)
Fraction stays constant
What stays constant when a drug is cleared by
first order kinetics is the "fraction"
eliminated with each pass of plasma through the
organ of clearance.
Whole body clearance and the elimination rate
constant are related, but emphasize different
aspects of the same processes.
Elimination Rate constant (Ke)
is the FRACTION of drug in the body that
is eliminated each unit of time, e.g.,
"fraction per hour".
Calculate Ke from CL & Vd: If
one knows the Vd of a drug and the CL(body), one
can calculate the FRACTION of the drug in the
body that is removed per unit time. This is the same
fraction as the fraction of "plasma
equivalent" that is completely
cleared of drug per unit time. This fraction is
the Ke.
Ke = CL(body) / Vd
(fraction/hour)
CL(body) = Ke x Vd
(volume/hour)
Diagram of the
Relationship among ER, Q, CL, Vd, and Ke.
There may be confusion about how one relates extraction ratio
and clearance with how much drug is actually eliminated during
any specific time interval. It should be noted that if a drug is
cleared by first order kinetics and if blood flow to the organ(s)
of elimination is constant, then CL and ER are both constant
regardless of how much drug there is in the plasma. However, THE
ACTUAL AMOUNT OF DRUG ELIMINATED DURING EACH INTERVAL OF AN HOUR
OR A MINUTE IS NOT CONSTANT. If the concentration of drug in the
plasma is rising, the amount of drug eliminated (e.g., mg/min) is
also rising. Vice versa, if the concentration of drug in the
plasma is falling (when drug absorbed per min is less than the
amount eliminated), then the amount of drug eliminated each
successive minute will also decrease.
To find the actual amount of drug eliminated each minute, one
must know the average concentration of drug in the arterial blood
to the organ of elimination during that minute. One then
multiplies that concentration times the CL to find the amount of
drug eliminated during that minute.
Amount of drug eliminated in the minute =
Average concentration in incoming plasma x CL
Is it possible to talk about the efficiency of an organ's
function in terms of its ability to "clear" a
drug from the circulation?
If one had tests to measure the functional capacity of an
organ, e.g., kidney, could one establish relationships
that would help one make "predictive dose
schedules" for certain drugs?
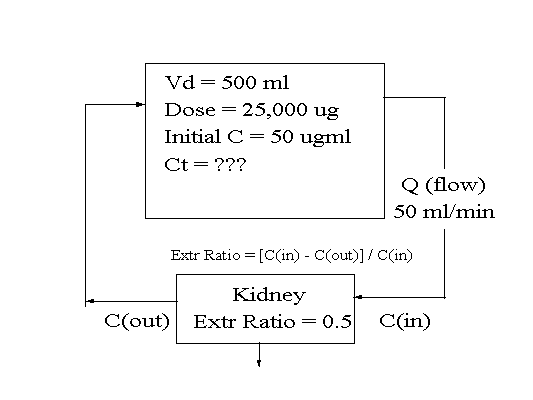
Assume that the Clearance of a drug by the kidney is 5
liters/hour and that the Vd is 50 liters. What FRACTION
of the Vd is completely "cleared" of drug per
hour? Could one point to the specific volume of blood
that had been freed of drug in the animal?
Assume the drug concentration at the beginning was 10
mg/L. How much drug would there be in the animal?
How much drug would there be in the animal at the end of
the first hour? ...the second hour?
Yes, it IS possible to talk about an organ's function in
terms of Clearance
Yes. In fact it is common to use such measures as
Creatinine Clearance as an index of the kidney's
filtration capacity. Many formulae have been developed to
adjust drug dosage, e.g., of aminoglycosides like
gentamicin, based on creatinine clearance.
If CL = 5 L/h and Vd = 50 L, then 5/50 or 1/10th of the
body fluid is cleared of drug per hour (0.1/h).
Given above parameters and an initial concentration of 10
mg/L, then the animal would contain 10 mg/L * 50 L = 500
mg of drug at the beginning.
If drug is cleared from 5 L/h, then 5 L * 10 mg/L = 50 mg
is eliminated in the first hour. Assuming volume loss was
negligible, then the new concentration is 450 mg/50 L = 9
mg/L. Thus 50 /500 or 1/10 of the drug was eliminated.
(0.1/h)
If drug is cleared from 5 L/h, then 5 L * 9 mg/L - 45 mg
eliminated in the second hour. This is 45/450 = 1/10 of
the drug present was eliminated the second hour (0.1/h).
At the same time 1/10th of the Vd was "cleared"
of drug in the same hour.
Note that the FRACTION of drug eliminated in each hour
was the same as the FRACTION of the Vd that was
"cleared" of drug in each hour. However, the
actual amount of drug eliminated decreased as the
concentration decreased.
This leads us to a discussion of elimination rate
constants and half-life: Ke
& t1/2
Ke, the elimination rate constant can be defined as the
fraction of drug in an animal that is eliminated per unit
of time, e.g., fraction/h.
Elimination half-life is the time required for the amount
of drug (or concentration) in the body to decrease by
half.
Although CL can be easily related to the function of a
specific organ, it is more difficult to get a
"minds-eye-view" of how fast a drug is removed
from the whole animal from CL. It is also not easy to
conceptualize what a Ke of 0.016 /h means. The
elimination "half-life" is better suited for
this because after 5 half-lives, approximately 97% of the
drug is gone.Therefore, it is useful to be able to
interconvert CL, Ke, and half-life.
CL, Ke, Vd, and Half-life are all inter-related as
follows:
0.693 is the natural logarithm of 2. Thus, half-life is
an arbitrary measure of drug elimination that is useful
for humans, but not easy to use in complex formulae.
Therefore, many tables contain both, but most formulae
require one to use Ke. You should be able to do this as
well as to inter-convert CL and Ke when given Vd.
The central
circulation is the reference point for all
bioavailability calculations. It is the only fluid that
has contact with the entire body.
Not all drug given to an animal per os (or other non-IV
routes) is absorbed into the central circulation.
Not all of an
administered dose is "available" to the sites
of action.
Bioavailability is defined as the fraction of drug
administered that reaches the central circulation,
i.e., the circulating post-portal venous blood.
By definition, bioavailability for a drug administered
intravenously is 100%. The "term" used is
"F". An F of 1.00 is equivalent to a
bioavailability of 100%.
One can find the "Area-Under-the-Curve" (AUC)
for a drug as a measure of the exposure of the animal to
the drug. In its simplest form, one plots the drug
concentration versus time and then adds up the areas of
successive trapezoids formed under the curve. This is the
so-called "trapezoidal rule".
A fun, simple way of determining the the relative drug
exposure would be to cut out the area under the plasma
concentration versus time plot and weigh it.
Find the area (weight!?) for the "test-route",
e.g., PO and compare it to the area (weight!?) of an
intravenous injection of the same drug dosage.
![full]](lg/pk01_19.gif){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 The central
circulation is the reference point for all
bioavailability calculations. It is the only fluid that
has contact with the entire body.
The central
circulation is the reference point for all
bioavailability calculations. It is the only fluid that
has contact with the entire body.  Not all of an
administered dose is "available" to the sites
of action.
Not all of an
administered dose is "available" to the sites
of action.  Graphic
depiction of determining F
Graphic
depiction of determining F